Research Overview
We are interested in understanding and harnessing photo-initiated charge and energy transfer in nanoscale systems, with a focus on nanoscale assemblies such as nanocrystal – organic molecule conjugates. This research has broad implications for technologies as diverse as artificial photosynthesis, bio-imaging, and quantum computation.
Our current interests can be broadly divided into three categories: photoexcited charge transfer from quantum dots for photocatalysis applications, surface functionalization of quantum dots for bio-imaging, and using nanocrystal-molecule systems to host photogenerated spin qubits. For background information and explanations of some of the jargon, please scroll down to the background section.
Photoexcited charge transfer from quantum dots
We seek to expand fundamental mechanistic understanding of photoexcited charge transfer processes in quantum dots. We are especially interested in experimental model systems composed of quantum dots covalently linked to molecular charge acceptors. One way to glean mechanistic information from charge transfer processes is to alter the free energies of either the charge donor or acceptor, and see how this affects charge transfer rates. In our quantum dot – molecular systems, we can tune the energy of the donor (quantum dot) by changing the size, composition, or surface passivation, and we can tune the energy of the acceptor (molecule) with synthetic attachment of electron donating and withdrawing functional groups. We also aim to explore the effect of solvent, temperature, physical distance, and a variety of other parameters in understanding charge transfer.
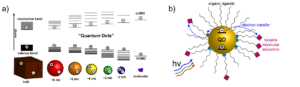
We plan to apply to results of this work to designing QD-molecular systems that can be used in biologically relevant contexts such as dynamic fluorescent sensors. Furthermore, we hope to extend our work to artificial photosynthesis applications by designing systems that can perform photocatalytic and photoelectrochemical carbon dioxide reduction.
Photogenerated spin qubits in nanoscale systems
Photoexcitation of an electron is followed closely in time by charge separation. The resultant charge separated state will consist of a radical cation and a radical anion until the charges recombine or cause a chemical reaction. However, while the charge separated state exists, it possesses unique properties. Specifically, the two unpaired electron spins associated with the two radicals (anion and cation) will be correlated and in a well-defined quantum state. These photogenerated charge states have been termed spin-correlated radical pairs or spin qubit pairs since they have the potential to find applications in quantum information technologies. We hope to use electron paramagnetic resonance techniques to detect these spin qubit pairs in the nanoscale systems described above.
Future interest: Charge and energy transfer in peptides
We are interested in using peptide and peptoid structures as scaffolds for exploring questions of how photogenerated charges and energy propagate on the nanoscale. The peptide structures will be both biologically inspired and artificial, and will be designed to support photoactive and redox active molecular moeities. Importantly, the automated nature of peptide synthesis will allow these structures to be highly tailorable by simply altering the sequence of residues. We hope to incorporate computational methods to simulate the charge and energy propagation in these systems to learn something about biological charge and energy transfer, and possibly apply this knowledge to creating artificial light harvesting systems.
Background information
Photophysical processes in quantum dots
What are photogenerated spin qubits?
Instrumentation
Fluorescence lifetime spectrometer
Time-correlated single photon counting lifetime spectrometer with laser diode excitation at specific wavelengths.
Steady state fluorimeter
Quantamaster 8075 with double excitation monochromators, an integrating sphere, and time-resolved phosphorescence capabilities.
Glovebox
A standard four arm glovebox for air-free manipulation and storage.
Gas Chromatograph
SRI Multiple Gas Analyzer for detecting and quantifying gaseous photocatalysis products such as carbon monoxide and hydrogen.
You must be logged in to post a comment.